「膵癌におけるATMの機能不全とそれに関連する化学療法」
日本の膵がん患者に多くみられる遺伝子変異に含まれるのが KRAS、ATM遺伝子などです。ここではATMの役割と化学療法との関係性について説明します。
▪要約
膵がんは、転移したがんを治療するための治療オプションがほとんどないまま、最も致死率の高い固形腫瘍の一つとして残っています。がん遺伝子パネル検査の利用により、膵がん患者の約25%で治療薬のある変異が同定されるようになりました。特にDNA損傷応答(DDR)遺伝子であり、がん細胞をDNA損傷剤やPARP阻害剤などのDNA損傷応答阻害剤に対して、より敏感にするものがわかってきました。ATM(Ataxia Telangiectasia Mutated)は最も一般的に変異するDDR遺伝子の一つで、膵がんの2-18%のケースで体細胞変異が、膵がん患者の1-34%で生殖細胞変異が同定されています。ATMは、細胞周期チェックポイントキナーゼで、多くの下流タンパク質の調節因子として、ゲノム安定性のためのDNA損傷応答を担当する複雑な役割を果たしています。ATM信号伝達経路の破綻は、他のDNA修復機構、特にATRやCHK1に依存することにつながり、ATM変異陽性の膵がんで下流タンパク質の阻害を利用した治療ターゲット化を可能にするかもしれません。このレビューでは、ATMの機能を詳細に説明し、膵がんにおけるATM欠損の現在のデータを検討し、ATM経路に関連する現在の臨床試験についても探求します。
▪イントロダクション
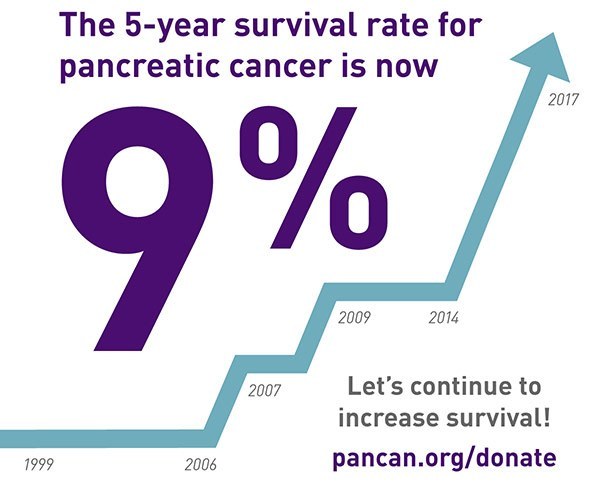
膵がん(膵がん)は、5年生存率が10%未満の最も致死率の高い固形腫瘍の一つであり続けています。米国では疾患の発症率は増加し、膵がんは2030年までにがんに関連する死亡者数の2番目の最も一般的な原因となると予測されています。高い死亡率は、診断時にほとんどの患者が局所進行または転移性疾患を持っているためです。残念なことに、新しい化学療法の改善にもかかわらず、生存期間中央値は1年未満です。潜在的な治療オプションを探るために包括的な遺伝子解析が行われており、最近のがん遺伝子パネル検査を利用した研究では、膵がんの約27%で治療薬のあるアクション可能な変異が同定されました。これらの発見は、他のいくつかの研究と一致しており、すべての研究で膵がんサンプルの17-48%でアクション可能なターゲットの遺伝子変異が発見されています。これらの大規模な次世代シーケンシングの取り組みに共通していることは、DNA損傷応答と修復(DDR)システムを調節する遺伝子の変異欠陥が同定されており、膵がんの17-25%で見られることです。
細胞周期中、DNAの60億以上の塩基対が複製されます。このようなゲノム複製は多くの侵害と複製ストレス因子にさらされるため、DNAの完全性を確保するために必要な基本的な応答と修復メカニズムに依存しています。さらに、がんを治療するために使用される化学療法、特に膵臓がんの化学療法は、特定の種類のDNA損傷を引き起こします。たとえば、プラチナなどのアルキル化剤やイリノテカンなどのトポイソメラーゼ阻害剤は二本鎖DNAの切断(DSBs)を引き起こします。一方、5-フルオロウラシルやジェムシタビンなどの抗代謝物質は単一塩基対の損傷を引き起こし、これが単一鎖DNAの切断を引き起こす可能性があります。DDRメカニズムの欠陥は治療のターゲットを明らかにし、研究を通してそのような欠陥を持つがんに対するFDA承認済みの治療法が開発されました。例えば、BRCA1およびBRCA2はゲノム完全性の維持に重要な役割を果たし、いずれの遺伝子の生殖細胞変異も乳がん、卵巣がん、膵臓がん、前立腺がんの発症リスクを増加します。ただし、BRCA1/2変異の存在は、トリプルネガティブ乳がんと膵がんの両方で白金系化学療法に対する感受性向上と全生存期間の改善を予測します。このDNA修復欠陥を利用することは、化学療法への感受性を向上させるだけでなく、PARPの阻害を通じてターゲット治療を可能にし、これにより単一鎖切断が蓄積し、DNA二重鎖の整合性が複製フォークで損なわれます。PARP阻害剤は、高度なBRCA1/2変異の卵巣がんと乳がんで無増悪生存期間を延長し、これらの疾患に対してFDA承認されています。また、PARP阻害剤への反応は、例えば50人の患者を対象としたトライアルで88%の反応率が見られた去勢抵抗性前立腺がんや、19および23人の患者を対象とした小規模な試験で、BRCA変異陽性膵臓がんでも16-22%の反応が見られます。
ATMもDDRで重要な役割を果たします。運動失調性毛細血管拡張症(ataxia telangiectasia)突然変異(ATM)遺伝子は、染色体11q 22–23に位置し、運動失調性毛細血管拡張症症候群の評価中に1995年に初めて同定されました。ATMの生殖細胞突然変異は、よく特徴化された症候群、および乳がん、膵臓がん、前立腺がんなどの発症リスクが増加します。ATM遺伝子の変異、生殖細胞または体細胞のいずれかで、膵がんの約6%まで見られ、したがってBRCA1/2よりも一般的なDDR変異を表す可能性があります。このレビューでは、ATMの機能を詳細に説明し、膵がんにおけるATMの不全の現在のデータを検討し、ATMの変異の治療的含意を検討します。
▪ATMの機能
ATM遺伝子は、DNA二本鎖切断(DSB)への応答および最終的にその修復に中心的な役割を果たすPI3K関連のセリン/スレオニンプロテインキナーゼをコードする66のエクソンから構成されています。この大きなタンパク質(350kDa)は、リン酸化の対象となるセリンまたはスレオニン残基を含み、親水性のターゲット領域に近いグルタミン酸アミノ酸が続きます。同様のポストトランスレーショナル修飾(PTM)のサイトは、運動失調性毛細血管拡張症およびRAD3関連(ATR)キナーゼ、DNAタンパク質キナーゼ(DNA-PK)タンパク質にも存在します。
ATMは、細胞内で重要な機能を果たしており、次の点を維持しています:
i) テロメアの長さの維持。
ii) 有糸分裂中のミトーシスにおけるミトーシススピンドル構造の維持。
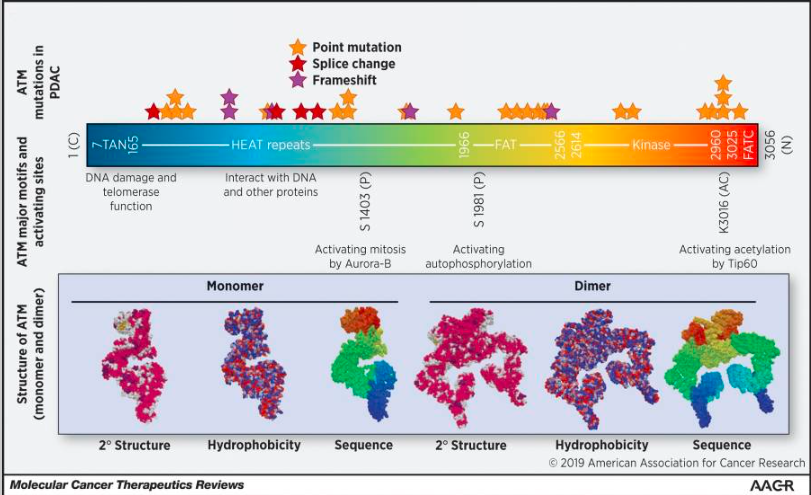
ただし、このレビューでは、ATMの中心的な役割に焦点を当て、がんにおける標的治療との関連性を含め、DDRプロセスでのATMの役割についてのみ議論します。図1に示されているように、損傷したDNAを修復するために、MRE11-RAD50-NBS1(MRN)複合体はDSBの主要なセンサーとして機能し、二つの切断端の物理的なブリッジを形成します。その後、ATMはNBS1(MRN複合体の一部)と直接相互作用し、NBS1のC末端がATMに存在するいくつかのHEATリピートに直接結合します。ATMの後続の活性化には、いくつかのPTMが必要とされると考えられています。たとえば、ATMはTIP60というヒストンアセチルトランスフェラーゼによるK3016のアセチル化を介して活性化されることが示されており、TIP60はATMをC末端のFATCドメインを認識することによって結合します。ATMはまた、S1981の自己リン酸化を必要とし、キナーゼドメインがFATドメインから分離し、キナーゼが活性化することを可能にします。これらの修飾は、DNA損傷に対する応答としてATMを非活性なホモ二量体から活性なモノマーに移行させます。このメカニズムは文献で支持されていますが、他の研究者からも疑問視されており、S1981および他のATM自己リン酸化イベントの役割を明確にするためにはさらなる研究が必要とされています。一旦活性化されると、ATMはDSB修復を実行し、アポトーシスやチェックポイント活性化などの正常な細胞周期プロセスも調節するため、複数の基質、タンパク質キナーゼ、センサータンパク質をリン酸化します。
▪DNA修復のためのATMの機能と他の関連経路
ATMは、MRN複合体によるNSB1とATMのHEATリピートとの直接の相互作用を介してDSBに引き寄せられます。次に、ATMは自己リン酸化とTIP60によるアセチル化を経て活性化され、この活性化によりATMは活性モノマー状態に分離できます。ATMモノマーは、その後、BRCA1とγ-H2AXを介してDNA修復のためのシグナルを送信できます。ATMはまた、直接リン酸化によるp53の活性化とCHK2およびMDM2を介した間接的な活性化を介して細胞周期の停止および/またはアポトーシスのシグナルも送ります。同時に、ATRは単一鎖DNA切断、DSBの切除、または複製ストレスによって引き起こされる単一鎖DNAの長いストレッチに引き寄せられます。PARP1は単一鎖切断の修復に重要な別の因子です。
ATMはDNA修復を開始するために必要なシグナル伝達に役立つ役割を果たします。したがって、ATMの異常はゲノム不安定性と悪性腫瘍につながる可能性があります。遺伝的および偶発的なATMの変異は、ATM遺伝子全体の機能領域にまたがって発生し、主にC末端に発生します。この領域はATMのアセチル化と活性化に関与しています。ATMタンパク質が機能不全の場合、DDRメカニズムが妨げられ、時間の経過とともに突然変異の蓄積が起こり、理論的には腫瘍形成のプロセスを開始する可能性があります。たとえば、ATMのゲルムラインポイント変異は乳がんのリスクを増加させ、特にS49CおよびS707P変異と関連しています。メラノーマ、前立腺がん、口咽頭がんは特にS49C変異と関連しています。一方、甲状腺または内分泌系のがんは一般的にS707P変異と関連しています。次世代シーケンシングは膵がんを含む多くの腫瘍タイプでソマティックATM変異も明らかにしました。
▪ATMの構造-機能ドメインと膵がんでの頻繁な変異
ATMには、モノマー、二量体のいずれかとしてのATMの機能にとって重要ないくつかの重要なドメインがあります。TANドメインは、テロメラーゼの機能とDSBへの勧誘にとって重要です。この勧誘は、ATMのHEATリピートとNBS1(MRN複合体の一部)との相互作用に依存しています(31,34)。FATドメインは通常、キナーゼ活性を二量体として抑制しますが、DNA損傷誘発型のS1981での自己リン酸化およびその後の二量体の分離により、キナーゼドメインが活性化します(36)。また、FATCドメインはTIP60との相互作用にとって重要であり、K3016でのTIP60によるATMのアセチル化はATMの活性化に必要です。Cbio-portalの膵がん患者におけるATM変異の変異解析(アクセス日:2019年1月21日)では、ATMにおける有意なクラスタリングやホットスポット変異は示されていませんが、患者数が少なかったです(N = 34)。
▪膵がんにおけるATM変異の無効化
多くの研究が膵がんにおけるTP53、KRAS、CDKN2A、SMAD4の変異の重要性を再確認しています。膵がんの遺伝的リスクは、BRCA1/BRCA2およびCDKN2Aのゲルマイン変異に大きく起因することが確立されており、家族性膵臓がんの7.4%で確認されました(N = 727)、またリンチ症候群(遺伝子:MLH1、MSH2、PMS2、MSH6の変異)に影響を受ける個人もいます。さらに、家族性膵がん患者の研究では、有害なATM変異は対照群よりも有意に高かったため、ATMの役割が示唆されています。この知識に基づいて、ATMのゲルマインおよびソマティック変異が識別され、感受性遺伝子変異のカタログに追加されました。
膵がんのゲノム特性の調査によれば、71のサンプルのうち37%がDNA修復遺伝子での変異を持っていることが明らかになりました。この重要な発見は、最近の大規模な膵がんプロファイリング研究でも確認され、640人の患者の50%でターゲットとなる変異が確認され、そのうち8.4%がBRCA1/2またはATM変異を発現していました。2015年に行われた国際がんゲノムコンソーシアムのレビューでは、膵がんの9〜18%でATM変異が確認され、591のサンプルの平均が12%でした。さらに、5万人の腫瘍サンプルの中で、同源組換え関連遺伝子変異の有病率を特定した大規模な人口研究も行われました。この研究では、ATM、ATRX、CHEK2の変異が1.3%のサンプルで確認されました。これらの変異は主に膵がんで特定されました。さらにゲノム研究は、膵がんでのATM、CHEK2、ATR変異の高い有病率を支持しています。総計5,234人の膵臓がん患者を対象とした文献検索によれば、膵がんにおけるATM変異(生殖細胞系または体細胞系)の総有病率は6.4%(範囲1〜34%)です。重要なことに、ATM生殖細胞系変異を所有する膵がん患者の約10%が、このうち44%の患者で体細胞系な二番目の変異が特定されました。この対立遺伝子の喪失(LOH)は、ATM生殖細胞変異を持つ患者から発生する腫瘍で頻繁に発生するようです(例:乳がんおよび膵がんで見られる)。しかし、LOHが治療感受性を伴う必要性(すなわち、プラチナとDDR阻害剤に対する感受性)は不確かです。
▪ATM欠損:治療の機会?
現在、DDRに基盤を持つ腫瘍細胞がDNA損傷剤、特にプラチナ剤に非常に敏感であることがよく確立されており、これは合成致死性(synthetic lethality)として知られる現象です。同様に、ATMの不活化変異もDNA損傷剤の存在下で合成致死性を引き起こす可能性があります。歴史的には、運動失調症状の患者は、染色体レベルで深刻な放射線感受性を示し、ATMとDNA修復の関連性を示唆しています。前臨床データは、ATMのノックダウンが放射線感受性を増加させることを示しています。研究室での追加の研究では、悪性細胞でのATM変異が細胞をプラチナ剤に対して敏感にすることを示し、プラチナ療法の外では、前臨床研究でもATM抑制はp53欠損マウス線維芽細胞をドキソルビシンに対して感受性を増加させることを示しました。以前の研究では、p53欠損細胞株でのATM欠損が5-FU感受性のわずかな増加を引き起こすことも示されています。最後に、ある研究では、マウスモデルでのATM変異陽性の膵がん細胞を利用し、PARP阻害剤のオラパリブやATR阻害剤のVE-822の治療によって、DSBの劇的な蓄積とin vitroおよびin vivoでの腫瘍細胞の生存率低下が示されました。著者は、ATM欠損をバイパスするための代替シグナル経路の補償、特に複製ストレス応答におけるATRの補償を指摘しました。したがって、ATR阻害は、深刻な有糸分裂損傷を促進するのに効果的であり、ジェムシタビンと組み合わせると効果が増強されました。
確認されたATM、ATR、またはCHEK2変異を持ち、特にオキサリプラチンベースの化学療法の有効性に焦点を当てた膵がんの臨床経験は非常に限られています。リアルタイム全エクソームシーケンシングを利用した1つの症例シリーズ(N=71)は、このような変異を持つ患者の大部分がオキサリプラチンベースの化学療法で一部応答または安定した状態を経験したことを示しました。この症例シリーズでは、ATM、ATR、またはCHEK2の変異を持つ患者の80%がオキサリプラチンベースの化学療法を受け、62.5%が最初のフォローアップスキャンで一部応答または安定した状態を示しました。別の小規模な研究(N=13)では、DDR変異腫瘍の患者におけるオキサリプラチンベースの化学療法療法への37.5%の応答率を示しました。DDR非変異腫瘍の患者と比較して、有意に長い無増悪生存期間も観察されました(20.8ヶ月対1.7ヶ月、p=0.049)。具体的には、30人の患者の中で、少なくとも1人の患者が5-FU、イリノテカン、オキサリプラチン(FOLFIRINOX)で約40ヶ月間の長期な部分的な応答を経験した、ATM変異を持つ患者が4人いました。ATM欠損腫瘍を対象とする多くの試験が現在進行中であり、特にPARP阻害剤を対象としています(Table 1)。
ATM、ATR、およびCHK1はすべてDNA損傷の解決に重要です(図1)。従って、DDRの基盤となる欠陥を利用し、別のキナーゼを阻害することで合成致死性を誘導するのは、がん細胞の死を引き起こす革新的な方法です。ATM、ATR、およびCHK1の小分子阻害剤の使用は、悪性細胞の急速で規制のない細胞分裂により、がん治療の有望な手法です。現在、ATMまたはATR阻害剤を単剤および化学療法との併用として使用した第Iおよび第II相の臨床試験が進行中です。
▪ATM阻害剤
ATMを阻害する最初の化合物はwortmanninでした、しかし、ATMを阻害するより新しい、より効力のある化合物が現在ではいくつか利用可能です。2004年に公表された新しい世代のATM阻害剤の1つは、KU55933でした。この化合物は、放射線照射後にATMの下流のリン酸化を阻害し、トポイソメラーゼ阻害剤であるエトポシド、カンプトテシン、ドキソルビシンに対する応答も向上させました。ATM阻害剤であるAZ31でも同様にトポイソメラーゼ阻害剤への感受性を示す研究が後に行われ、PDXモデルで耐性腫瘍においてイリノテカンの効果を増強することが示されました。KU60019は、KU55933の改良アナログとして2009年に導入された別の化合物であり、初期の研究ではKU60019が強力な放射線感作剤であることが示されました。この分子は現在、別の既知の感作剤であるCK2の蛋白質キナーゼを阻害するCX4945との組み合わせで、クリアセル腎細胞癌で研究されており、この組み合わせはCK2とATM阻害剤が腎癌で高度に相乗的であることが化合物スクリーニングで示されたことに基づいています。興味深いことに、CK2阻害剤が同等のATM存在/不在のマウス細胞株で試験された際、AKT1やBIDなどのDNA修復の下流効果においてはほとんど差がなかったが、CK2阻害剤で治療されたATM存在/不在の細胞株の全体的な生存性は評価されていなかった(76)。ATM阻害剤と併用して介入の効果をATM不全の腫瘍を持つ患者と比較する際、ATM不全が慢性的なATMの機能喪失に適応した可能性があるため、結果は異なる可能性があります。逆に、他の介入はATM阻害剤と相乗的であり、ATM不全の患者ではより効力がある可能性があります。
ATM阻害剤の前臨床的な探索を超えて、他の治療法と併用して調査されている2つのATM阻害剤が現在臨床試験中であり、(表1参照)他の治療法との併用で研究されています。AZD0156は、口から摂取するATM阻害剤であり、現在はolaparibまたはFOLFIRIとの併用で臨床試験中です。これらの組み合わせは、以前に述べたように、ATM阻害剤がPARP阻害剤に対する細胞の感受性を高めることが示されており、また5-FUおよびイリノテカンに対しても感受性が高まることが示されています。もう1つの口から摂取するATM阻害剤であるAZD1390は、血液脳関門を通過するATM阻害剤であり、放射線との併用で研究されており、放射線はATM不全のがんでより効果的であることが示されています。この試験の潜在的な副作用を考慮する際、ATMのノックアウトは、がん細胞とは異なり、健康な組織でのATMのノックアウトは放射線感受性を低下させないことが示されました。ATM阻害剤が臨床でさらに探索されるにつれて、特に他の治療法との併用でATM阻害剤の副作用を監視することが重要です。
ATM不全の腫瘍は以前にPARP阻害剤に対する高い感受性が示されています、および124人の患者の臨床試験では、オラパリブとパクリタキセルの投与がATM活性が低い患者で全生存期間を増加させるのに効果的であることが示されました(HR、0.35; 80% CI、0.22 to 0.56; P = .002; median OS、not reached v 8.2 months)。残念ながら、525人の患者を対象としたその後の第III相試験では、ATM不全の腫瘍を富化させず、否定的な結果となりました。それにもかかわらず、ATM不全の腫瘍を少なくとも一部対象とするPARP阻害剤に基づく治療に関する多くの進行中の試験があります(表1参照)。
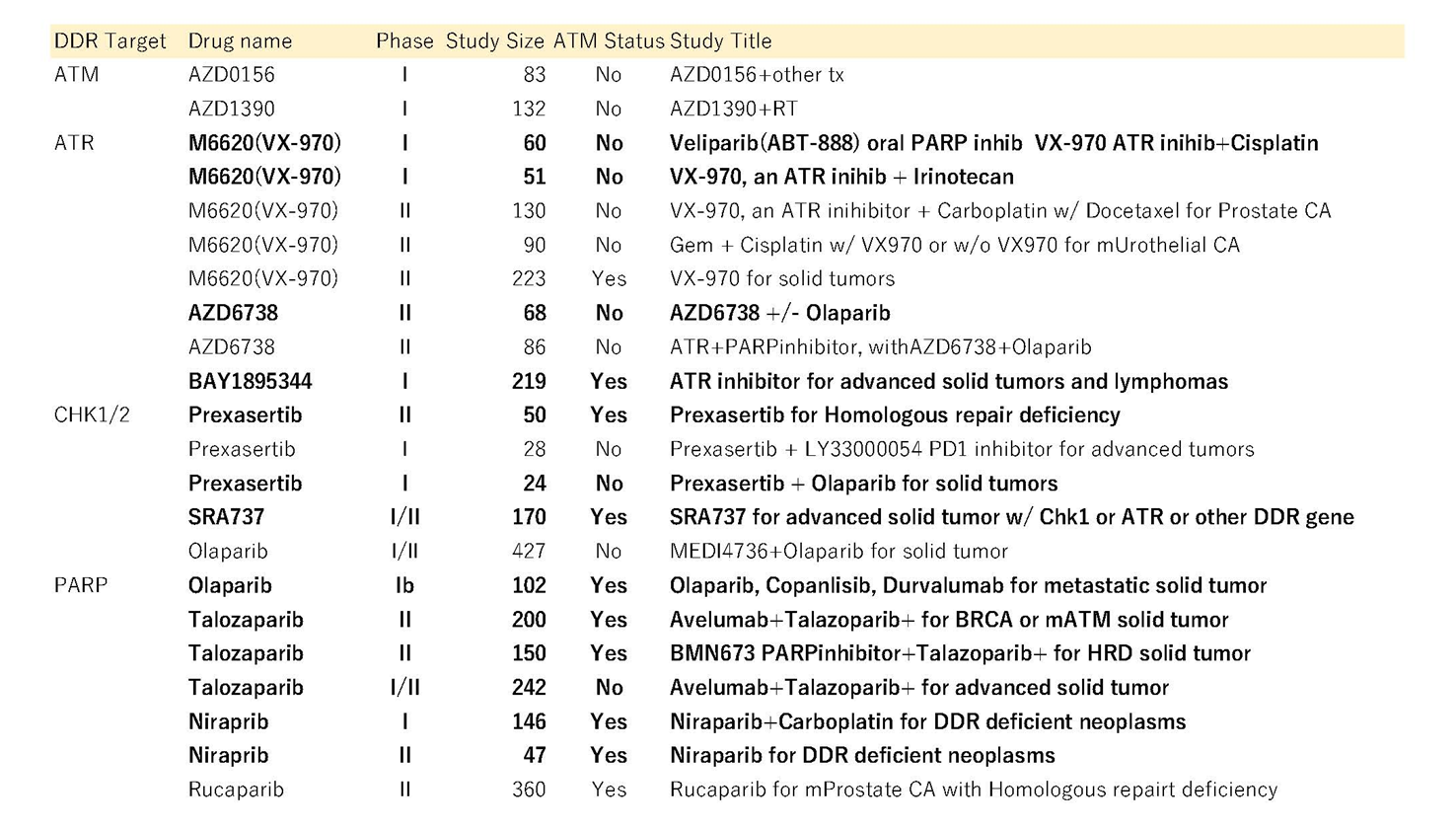
表1 ATM関連の臨床試験。
この表は、現在進行中のATM欠損腫瘍を直接または間接的に標的とする臨床試験を要約しています。これらの試験は、2019年3月2日にclinicaltrials.govでの検索から収集されました。膵臓がん患者を受け入れる可能性のある臨床試験は太字で表示されています。
▪ATR阻害剤
ATRは主に一本鎖DNAの切断に反応し、修復するフォスホイノシチド3キナーゼ関連プロテインキナーゼであり、DSBに反応するATMとDNA-PKと機能的な配列を共有しています。ATMによる上流タンパク質のリン酸化とT1989サイトでの自己リン酸化は、ATRの活性を刺激し、またATR活性を促進するATR活性化ドメインを含むTopBP1も同様です。ATRキナーゼはさまざまな細胞ストレスに反応し、複製中にDNAの完全性を維持し、増殖する細胞の生存に不可欠です。急速に分裂するがん細胞では、複製のストレスが高度であり、それにより、ATR活性の抑制が複製のストレスをさらに増大させ、細胞死につながることが、前臨床研究で示されています。さらに、通常の分裂細胞はDNA修復の支援にATM依存性の経路を利用しますが、がん細胞は通常、ATM/p53シグナリングが不足していることが多く、生存にはATR経路だけに依存する場合があります。これは、がんの遺伝子工学的に変更されたマウスモデルで示され、ATR発現の90%の遺伝子削減は、正常な組織にほとんどの副作用を持たずに線状肉腫および急性骨髄性白血病の発症を抑制することが確認されました。この研究は、ATR阻害の腫瘍選択性を確認しました。さらに、ATRの阻害は、放射線療法と化学療法に対して腫瘍細胞の感受性を選択的に高めますが、正常細胞には影響を与えません。
したがって、ATMの体細胞変異を持つ膵がんにおいて、ATMの機能の不足はATRへの高い依存性を引き起こす可能性があるため、ATR阻害剤はがん細胞の死を大幅に促進できる可能性があります。ATR阻害剤であるVE-821は、がん細胞を化学療法に対して感受性を高めますが、正常細胞には影響しません。これらの効果はATM不全の細胞で相乗的でした。別のATR阻害剤であるAZD6738は、ATMの機能の不全な胃細胞でDNA損傷の蓄積、S期の停止、およびアポトーシスを引き起こしますが、機能的なATMを持つ細胞には影響しません。同様の前臨床研究は、膵がんおよび肺腺癌細胞株におけるATR阻害とATMの合成致死性を示唆し、ATRへのアクション可能な分子依存性を再確認しています。VE-822は、ATM不全の食道扁平細胞においてシスプラチンとの相乗作用を示す可能性も示されています。シスプラチンおよびジェムシタビンの細胞毒性の増強効果は、ATM不全の非小細胞肺がん細胞でもAZD6738によって見られます。ATR阻害剤を用いた治療のいくつかの進行中の試験があり、それらは表1にリストされています。
▪CHK1およびCHK2阻害剤
ATRによって下流で活性化されるのは、チェックポイントキナーゼ1(CHK1)経路です。CHK1はゲノムストレスに対するCDC25Aのプロテアソーマル分解を促進します。ATR、CHK1、CDC25Aの結合活性は、細胞周期の停止とDNAフォークでの複製ストレスの安定化をもたらします。この複合体の阻害は、フォーク進行の速度の減少、S期細胞での大規模なフォーク崩壊、最終的には細胞死につながります。 CHK1阻害剤であるMK8776とLY2603618をジェムシタビンベースのケモラジオ療法と組み合わせて使用した前臨床研究では、PDAC細胞のアポトーシスを誘導する協力的な効果が示されました。 CHK1阻害剤であるPF-477736を含むジェムシタビン、チェックポイントキナーゼ1(CHK1)阻害剤、ルテチウム-177ラベルの抗EGFR抗体の組み合わせは、患者由来のゼノグラフトで広範なDNA損傷、アポトーシス、および腫瘍変性を引き起こしました。 チェックポイントキナーゼ1(CHK1)およびチェックポイントキナーゼ2(CHK2)阻害剤を使用した前臨床研究は、ジェムシタビンとの組み合わせで膵臓細胞でアポトーシスを増加させました。 CHK1/2阻害剤の臨床試験も表1に示されています。
▪結語
現在、膵がん患者向けに承認された標的治療法はありません。ただし、膵臓腺癌のゲノムプロファイリングは治療に関連する遺伝子変異を明らかにし、17-25%の膵臓がんがDDR経路の変異を保有しています。PARP阻害剤を含むDNA損傷応答を阻害する療法は、特にDDR欠損のがんで高い効果を示しており、これはBRCA1/2変異のがんで確立されています。ただし、DNA損傷応答は非常に複雑なプロセスであり、いくつかの重なり合う経路を含んでいます。特定のDDR変異に応じて、最適な治療法が異なる可能性があると仮定するのは合理的です。PARP阻害剤は、BRCA1/2またはPALB2変異を有するPDACで初期の有望な結果を示していますが、PDACで最も一般的にDDR遺伝子が変異するのは実際にはATMです。 ATM不全の膵がんにおいてどのDDRターゲット治療法が最も効果的であるかを探求することが今後の重要な課題となります。治療のブレークスルーに伴うように、DDRパスウェイの複雑さを探求する未来においても、ATM/ATR/CHK1を対象とした治療において生じるかもしれない補償的および耐性メカニズムの理解が必要です。
ATM欠損は、膵がんの放射線(96)およびオキサリプラチンなどの従来の治療法の要素に対する感受性を提供する可能性があります。しかし、免疫療法を含む新興のターゲット治療戦略(97)も、おそらくATM不全腫瘍にとってより適しているでしょう。たとえば、機械的には、PARP阻害剤がATM不全の腫瘍の治療に効果的であると考えられます。しかし、DNA損傷を誘発する必要がある点も考慮することが重要です。前述のように、多くの進行中の臨床試験がありますが、ATM不全が一般的であり、未だに十分な治療法が存在しないPDACにおいて、臨床試験が理想的であるでしょう。
さらに、最近の遺伝学的研究では、特定のATM遺伝子型ががんを含むさまざまな疾患への感受性と相関することが明らかになっており、膵がんの早期検出、サブタイプ分類、および治療に関する臨床情報を提供する可能性があります。これらの遺伝学的研究は、患者で観察されるATM病原性遺伝子型を評価するために役立つか、または評価されるかもしれません。これらの遺伝学的研究は、膵がんの進行モデルにおけるATMのさまざまな役割(すなわちEMT、遺伝的不安定性、および転移)を特定した遺伝子工学的に調整されたマウスモデルで評価されるべきです。今後、研究コミュニティは、上記で議論した新規の薬剤および組み合わせ療法を、最適に合った対象指向型治療戦略で患者に提供できるように、これらの同所性のin vivoモデルで評価するべきです。
ーーーーーーーーーーーーーーーーーーーーーーーーーーー
<免責事項>この記事は、膵臓がんに関連した最新のサイエンスを紹介する目的で書かれています。特定の治療法や薬の使用を推奨するものではありません。ご自身の病状については、担当医とよく話し合ってください。このウェブサイトの情報を利用して生じた結果についてPanCANJapanは一切責任を負うことができませんのでご了承ください。
編集注:本記事は本記事はATM欠損と関連する化学療法について紹介するために作成されました。
NIH National Library of Medicineにより出版された英語文献を自動翻訳したものです。
正確な内容を把握されたい方は、英語の原文をお読みになることをお薦めします。
SOURCE;
Mol Cancer Ther. Author manuscript; available in PMC 2020 May 1.
Published in final edited form as:
Mol Cancer Ther. 2019 Nov; 18(11): 1899–1908.
doi: 10.1158/1535-7163.MCT-19-0208
PMCID: PMC6830515
NIHMSID: NIHMS1539120
PMID: 31676541
ATM Dysfunction in Pancreatic Adenocarcinoma and Associated Therapeutic Implications
Samantha A. Armstrong,1 Christopher W. Shultz,2 Ariana Azimi-Sadjadi,1 Jonathan R. Brody,2 and Michael J. Pishvaian3
Author information Copyright and License information PMC Disclaimer